Brings SummarizedExperiment to the tidyverse!
website: stemangiola.github.io/tidySummarizedExperiment/
Another nice introduction by carpentries-incubator.
Please also have a look at
- tidySingleCellExperiment for tidy manipulation of SingleCellExperiment objects
- tidyseurat for tidy manipulation of Seurat objects
- tidybulk for tidy analysis of RNA sequencing data
- nanny for tidy high-level data analysis and manipulation
- tidygate for adding custom gate information to your tibble
- tidyHeatmap for heatmaps produced with tidy principles
Introduction
tidySummarizedExperiment provides a bridge between Bioconductor SummarizedExperiment [@morgan2020summarized] and the tidyverse [@wickham2019welcome]. It creates an invisible layer that enables viewing the Bioconductor SummarizedExperiment object as a tidyverse tibble, and provides SummarizedExperiment-compatible dplyr, tidyr, ggplot and plotly functions. This allows users to get the best of both Bioconductor and tidyverse worlds.
Functions/utilities available
| SummarizedExperiment-compatible Functions | Description |
|---|---|
all |
After all tidySummarizedExperiment is a SummarizedExperiment object, just better |
| tidyverse Packages | Description |
|---|---|
dplyr |
Almost all dplyr APIs like for any tibble |
tidyr |
Almost all tidyr APIs like for any tibble |
ggplot2 |
ggplot like for any tibble |
plotly |
plot_ly like for any tibble |
| Utilities | Description |
|---|---|
as_tibble |
Convert cell-wise information to a tbl_df
|
Installation
if (!requireNamespace("BiocManager", quietly=TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("tidySummarizedExperiment")From Github (development)
devtools::install_github("stemangiola/tidySummarizedExperiment")Load libraries used in the examples.
Create tidySummarizedExperiment, the best of both worlds!
This is a SummarizedExperiment object but it is evaluated as a tibble. So it is fully compatible both with SummarizedExperiment and tidyverse APIs.
pasilla_tidy <- tidySummarizedExperiment::pasilla It looks like a tibble
pasilla_tidy## # A SummarizedExperiment-tibble abstraction: 102,193 × 5
## [90m# Transcripts=14599 | Samples=7 | Assays=counts[39m
## .feature .sample counts condition type
## <chr> <chr> <int> <chr> <chr>
## 1 FBgn0000003 untrt1 0 untreated single_end
## 2 FBgn0000008 untrt1 92 untreated single_end
## 3 FBgn0000014 untrt1 5 untreated single_end
## 4 FBgn0000015 untrt1 0 untreated single_end
## 5 FBgn0000017 untrt1 4664 untreated single_end
## 6 FBgn0000018 untrt1 583 untreated single_end
## 7 FBgn0000022 untrt1 0 untreated single_end
## 8 FBgn0000024 untrt1 10 untreated single_end
## 9 FBgn0000028 untrt1 0 untreated single_end
## 10 FBgn0000032 untrt1 1446 untreated single_end
## # … with 40 more rowsBut it is a SummarizedExperiment object after all
assays(pasilla_tidy)## List of length 1
## names(1): countsTidyverse commands
We can use tidyverse commands to explore the tidy SummarizedExperiment object.
We can use slice to choose rows by position, for example to choose the first row.
## # A SummarizedExperiment-tibble abstraction: 1 × 5
## [90m# Transcripts=1 | Samples=1 | Assays=counts[39m
## .feature .sample counts condition type
## <chr> <chr> <int> <chr> <chr>
## 1 FBgn0000003 untrt1 0 untreated single_endWe can use filter to choose rows by criteria.
## # A SummarizedExperiment-tibble abstraction: 58,396 × 5
## [90m# Transcripts=14599 | Samples=4 | Assays=counts[39m
## .feature .sample counts condition type
## <chr> <chr> <int> <chr> <chr>
## 1 FBgn0000003 untrt1 0 untreated single_end
## 2 FBgn0000008 untrt1 92 untreated single_end
## 3 FBgn0000014 untrt1 5 untreated single_end
## 4 FBgn0000015 untrt1 0 untreated single_end
## 5 FBgn0000017 untrt1 4664 untreated single_end
## 6 FBgn0000018 untrt1 583 untreated single_end
## 7 FBgn0000022 untrt1 0 untreated single_end
## 8 FBgn0000024 untrt1 10 untreated single_end
## 9 FBgn0000028 untrt1 0 untreated single_end
## 10 FBgn0000032 untrt1 1446 untreated single_end
## # … with 40 more rowsWe can use select to choose columns.
## # A tibble: 102,193 × 1
## .sample
## <chr>
## 1 untrt1
## 2 untrt1
## 3 untrt1
## 4 untrt1
## 5 untrt1
## 6 untrt1
## 7 untrt1
## 8 untrt1
## 9 untrt1
## 10 untrt1
## # … with 102,183 more rowsWe can use count to count how many rows we have for each sample.
## # A tibble: 7 × 2
## .sample n
## <chr> <int>
## 1 trt1 14599
## 2 trt2 14599
## 3 trt3 14599
## 4 untrt1 14599
## 5 untrt2 14599
## 6 untrt3 14599
## 7 untrt4 14599We can use distinct to see what distinct sample information we have.
## # A tibble: 7 × 3
## .sample condition type
## <chr> <chr> <chr>
## 1 untrt1 untreated single_end
## 2 untrt2 untreated single_end
## 3 untrt3 untreated paired_end
## 4 untrt4 untreated paired_end
## 5 trt1 treated single_end
## 6 trt2 treated paired_end
## 7 trt3 treated paired_endWe could use rename to rename a column. For example, to modify the type column name.
## # A SummarizedExperiment-tibble abstraction: 102,193 × 5
## [90m# Transcripts=14599 | Samples=7 | Assays=counts[39m
## .feature .sample counts condition sequencing
## <chr> <chr> <int> <chr> <chr>
## 1 FBgn0000003 untrt1 0 untreated single_end
## 2 FBgn0000008 untrt1 92 untreated single_end
## 3 FBgn0000014 untrt1 5 untreated single_end
## 4 FBgn0000015 untrt1 0 untreated single_end
## 5 FBgn0000017 untrt1 4664 untreated single_end
## 6 FBgn0000018 untrt1 583 untreated single_end
## 7 FBgn0000022 untrt1 0 untreated single_end
## 8 FBgn0000024 untrt1 10 untreated single_end
## 9 FBgn0000028 untrt1 0 untreated single_end
## 10 FBgn0000032 untrt1 1446 untreated single_end
## # … with 40 more rowsWe could use mutate to create a column. For example, we could create a new type column that contains single and paired instead of single_end and paired_end.
## # A SummarizedExperiment-tibble abstraction: 102,193 × 5
## [90m# Transcripts=14599 | Samples=7 | Assays=counts[39m
## .feature .sample counts condition type
## <chr> <chr> <int> <chr> <chr>
## 1 FBgn0000003 untrt1 0 untreated single
## 2 FBgn0000008 untrt1 92 untreated single
## 3 FBgn0000014 untrt1 5 untreated single
## 4 FBgn0000015 untrt1 0 untreated single
## 5 FBgn0000017 untrt1 4664 untreated single
## 6 FBgn0000018 untrt1 583 untreated single
## 7 FBgn0000022 untrt1 0 untreated single
## 8 FBgn0000024 untrt1 10 untreated single
## 9 FBgn0000028 untrt1 0 untreated single
## 10 FBgn0000032 untrt1 1446 untreated single
## # … with 40 more rowsWe could use unite to combine multiple columns into a single column.
## # A SummarizedExperiment-tibble abstraction: 102,193 × 4
## [90m# Transcripts=14599 | Samples=7 | Assays=counts[39m
## .feature .sample counts group
## <chr> <chr> <int> <chr>
## 1 FBgn0000003 untrt1 0 untreated_single_end
## 2 FBgn0000008 untrt1 92 untreated_single_end
## 3 FBgn0000014 untrt1 5 untreated_single_end
## 4 FBgn0000015 untrt1 0 untreated_single_end
## 5 FBgn0000017 untrt1 4664 untreated_single_end
## 6 FBgn0000018 untrt1 583 untreated_single_end
## 7 FBgn0000022 untrt1 0 untreated_single_end
## 8 FBgn0000024 untrt1 10 untreated_single_end
## 9 FBgn0000028 untrt1 0 untreated_single_end
## 10 FBgn0000032 untrt1 1446 untreated_single_end
## # … with 40 more rowsWe can also combine commands with the tidyverse pipe %>%.
For example, we could combine group_by and summarise to get the total counts for each sample.
## # A tibble: 7 × 2
## .sample total_counts
## <chr> <int>
## 1 trt1 18670279
## 2 trt2 9571826
## 3 trt3 10343856
## 4 untrt1 13972512
## 5 untrt2 21911438
## 6 untrt3 8358426
## 7 untrt4 9841335We could combine group_by, mutate and filter to get the transcripts with mean count > 0.
## # A tibble: 86,513 × 6
## # Groups: .feature [12,359]
## .feature .sample counts condition type mean_count
## <chr> <chr> <int> <chr> <chr> <dbl>
## 1 FBgn0000003 untrt1 0 untreated single_end 0.143
## 2 FBgn0000008 untrt1 92 untreated single_end 99.6
## 3 FBgn0000014 untrt1 5 untreated single_end 1.43
## 4 FBgn0000015 untrt1 0 untreated single_end 0.857
## 5 FBgn0000017 untrt1 4664 untreated single_end 4672.
## 6 FBgn0000018 untrt1 583 untreated single_end 461.
## 7 FBgn0000022 untrt1 0 untreated single_end 0.143
## 8 FBgn0000024 untrt1 10 untreated single_end 7
## 9 FBgn0000028 untrt1 0 untreated single_end 0.429
## 10 FBgn0000032 untrt1 1446 untreated single_end 1085.
## # … with 86,503 more rowsPlotting
my_theme <-
list(
scale_fill_brewer(palette="Set1"),
scale_color_brewer(palette="Set1"),
theme_bw() +
theme(
panel.border=element_blank(),
axis.line=element_line(),
panel.grid.major=element_line(size=0.2),
panel.grid.minor=element_line(size=0.1),
text=element_text(size=12),
legend.position="bottom",
aspect.ratio=1,
strip.background=element_blank(),
axis.title.x=element_text(margin=margin(t=10, r=10, b=10, l=10)),
axis.title.y=element_text(margin=margin(t=10, r=10, b=10, l=10))
)
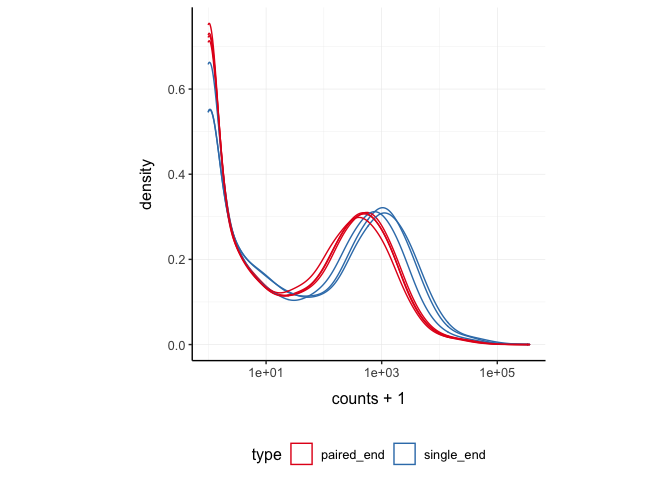
)We can treat pasilla_tidy as a normal tibble for plotting.
Here we plot the distribution of counts per sample.
pasilla_tidy %>%
tidySummarizedExperiment::ggplot(aes(counts + 1, group=.sample, color=`type`)) +
geom_density() +
scale_x_log10() +
my_theme